Ein Restrisiko bleibt immer bestehen: Risikomanagement für Medizinprodukte10 | 01 | 20
 Ich gebe es zu, es gibt bestimmte Prinzipien aus dem Qualitätsmanagement, die wende ich auch in meinem Privatleben an. Allerdings folgen meine persönlichen Dokumente keinem ausgeklügeltem Prüf- und Freigabesystem und auch die Lagerhaltung in meinem Keller unterliegt einer anderen Definition von chaotischer Lagerhaltung als man sie aus der Logistik kennt. Reklamationen bei meinen Kochkünsten werden auf keinen Fall systematisch erfasst und meinen Internetanbieter hätte ich bei funktionierendem Lieferantenmanagement mit Sicherheit bereits gewechselt. Aber den Gedanken des risikobasierten Ansatzes, der sich wie ein roter Faden durch sämtliche Anforderungen für Medizinprodukte zieht, verfolge ich auch privat. Zum Beispiel beim Kauf eines neuen Fahrradschlosses.
Ich gebe es zu, es gibt bestimmte Prinzipien aus dem Qualitätsmanagement, die wende ich auch in meinem Privatleben an. Allerdings folgen meine persönlichen Dokumente keinem ausgeklügeltem Prüf- und Freigabesystem und auch die Lagerhaltung in meinem Keller unterliegt einer anderen Definition von chaotischer Lagerhaltung als man sie aus der Logistik kennt. Reklamationen bei meinen Kochkünsten werden auf keinen Fall systematisch erfasst und meinen Internetanbieter hätte ich bei funktionierendem Lieferantenmanagement mit Sicherheit bereits gewechselt. Aber den Gedanken des risikobasierten Ansatzes, der sich wie ein roter Faden durch sämtliche Anforderungen für Medizinprodukte zieht, verfolge ich auch privat. Zum Beispiel beim Kauf eines neuen Fahrradschlosses.
Die Welt ist voller Risiken, die kontrolliert werden müssen
Für Medizinprodukte gibt eine eigene Norm für das Management von Risiken. DIN EN ISO 14971 beschreibt Anforderungen und mögliche Verfahren im Rahmen einer Risikoanalyse. Hersteller müssen Risiken identifizieren, bewerten und darüber entscheiden, ob diese akzeptabel sind. Nicht-akzeptable Risiken erfordern Kontrollmaßnahmen. Dies kann durch gezielte Eingaben an das Design erfolgen, aber auch durch Schutzmaßnahmen und besondere Maßnahmen während der Herstellung. In letzter Instanz erfolgt eine Risikokontrolle durch das Bereitstellen von Informationen zur Sicherheit an den Benutzer. Verbleibende Restrisiken müssen ins Verhältnis zum Nutzen für den Patienten gesetzt werden. Dass der gesamte Prozess des Risikomanagements geplant werden muss, ahnt ein guter „Qualitäter“ sicherlich sofort.
Analyse von Risiken
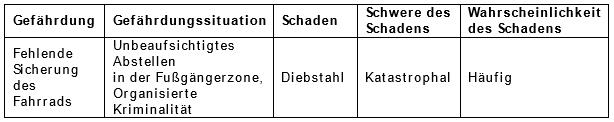
Ein Risiko ergibt sich aus der Kombination von Wahrscheinlichkeit P und Schwere S eines Schadens. Ein Schaden entsteht mit einer gewissen Wahrscheinlichkeit P aus einer Gefährdung, bzw. Gefährdungssituationen. Das klingt kompliziert, lässt sich jedoch am Beispiel des Fahrraddiebstahls ganz gut veranschaulichen. Eine fehlende Sicherung meines Fahrrads (Gefährdung) führt nicht automatisch zum Diebstahl (Schaden), welche für mich sehr schmerzhaft wäre (Schwere des Schadens). Erst durch das Abstellen in der Fußgängerzone oder durch die Anwesenheit organisierter Banden (Gefährdungssituation) entsteht das Risiko. Da der Diebstahl des Fahrrads in der genannten Gefährdungssituation sehr wahrscheinlich ist, klassifiziere ich ihn als häufig. Die Schwere des Schadens ist hier maximal, deswegen wird er als katastrophal bewertet. Für mich ist das Risiko nicht akzeptabel. Die Klassifizierung des Risikos obliegt dem Hersteller, die Norm bietet jedoch konkrete Vorschläge zur Umsetzung an.
In einer Risikoanalyse nach DIN EN ISO 14971 würde dieses Szenario wie folgt dokumentiert werden:
Risikokontrolle und Risiko-Nutzen-Bewertung
Kontrollieren lässt sich das Risiko durch den Kauf eines zusätzlichen Schlosses. Außerdem sollte das Fahrrad am besten an einem fest installierten Fahrradständer befestigt werden. Da in früheren Lebensphasen gezeigt wurde, dass selbst gute Schlösser nicht vor Entwendung durch organisierte Banden schützen, lässt sich das Risiko nur für die Spontanentwendung mindern. Ich habe dieses jedoch soweit wie möglich gemindert. Das verbleibende Restrisiko durch organisierte Kriminalität muss ich akzeptieren. Die Norm fordert mich weiterhin auf, das verbleibende Risiko im Verhältnis zum Nutzen zu bewerten. Der Nutzen des Fahrrads überwiegt für mich und es besteht keine Notwendigkeit, den Besitz des Fahrrads infrage zu stellen.
Es gibt allgemeine und spezifische Risken für bestimmte Produktgruppen
Welche Risiken innerhalb der Risikoanalyse betrachtet werden müssen, hängt vom Produkt ab. Grundsätzlich gibt die DIN EN ISO 14971 eine gute Übersicht über relevante Aspekte. Doch auch andere Normen geben Hinweise darüber, welche Fragen im Rahmen der Risikoanalyse gestellt werden sollten. So müssen aktive Medizinprodukte gemäß DIN EN 60601-1 bewertet werden. Das beinhaltet unter anderem die Analyse von Risiken, die verbunden sind mit dem Auftreten eines Erstfehlers oder dem Eindringen von Flüssigkeiten in das Produkt. Außerdem gibt die EN 62366-1 Anhaltspunkte zur Identifizierung von benutzungsbezogenen Risiken. Die EN ISO 10993-1 beschreibt weiterhin Anforderungen an die Biokompatibilitätsbewertungen, um beispielsweise das Auftreten von Allergien beim Patienten zu verhindern.
Die Risikoanalyse steht am Anfang jeder Produktentwicklung
Letztendlich beginnt jede Entwicklung eines Medizinprodukts mit einer qualitativen Analyse möglicher Risiken, insbesondere für den Patienten. Bevor es einen vorzeigefähigen Prototyp gibt, sollte es eine dokumentierte Risikoanalyse geben. Das ist essentiell, denn nur so kann bereits in frühen Phasen der Entwicklung verhindert werden, dass die Benutzung eines Medizinprodukts zu einer nicht akzeptablen Gefährdung des Patienten führen kann oder Herstellungsprozesse nicht realisierbar sind. Spätere Designänderungen sind mühsam, zeitaufwendig und letztendlich teuer. Das heißt ein frühzeitiges systematisches Bewerten hilft nicht nur der Optimierung des Produkts, sondern auch der Vermeidung späterer Kosten.
Der risikobasierte Ansatz führte bei mir zu der Entscheidung nur noch ein mittelpreisiges Schloss zu kaufen und mein Rad im Keller einzuschließen, wenn ich es nicht fahre. Klingt nach gesundem Menschenverstand. Letztendlich meint der risikobasierte Ansatz auch genau das.
Revision der ISO 14971
Vor kurzem erschien eine Revision der Risikomanagementnorm für Medizinprodukte als ISO 14971:2019. Das grundsätzliche Verfahren zur Risikobewertung ändert sich damit nicht, aber einige Besonderheiten bleiben dennoch zu berücksichtigen. Neu hinzugekommen ist unter anderem die Option Information for Training als Risikokontrollmaßnahme. Der Begriff des Nutzens für den Patienten wurde weiter spezifiziert, außerdem wurde der Begriff des State of the Art eingeführt. Die Bewertung des Verhältnisses von Risiko zu Nutzen für den Patienten rückt damit in einen engeren Fokus. Ein Review der Risikomanagementakte muss nun erfolgen und dokumentiert werden. Außerdem ergänzen sich die Vorgaben an Post-Market-Aktivitäten mit der MDR (Medical Device Regulation; Europäische Verordnung über Medizinprodukte). Viele Anhänge wurden in die TR/ISO 24971, die zeitgleich überarbeitet wurde, verwiesen. Ob die Norm im Zusammenhang mit der MDR harmonisiert wird und wie sie dazu dienen kann die grundlegenden Sicherheits- und Leistungsanforderungen nach MDR (2017/745) zu erfüllen, werden wir sicherlich bald von der Europäischen Kommission erfahren.
Referenzen:
- DIN EN ISO 14971 Medizinprodukte – Anwendung des Risikomanagements auf Medizinprodukte
- DIN EN 60601-1 Medizinische elektrische Geräte; Teil 1: Allgemeine Festlegungen für die Sicherheit einschließlich der wesentlichen Leistungsmerkmale
- DIN EN 62366-1 Medizinprodukte – Teil 1: Anwendung der Gebrauchstauglichkeit auf Medizinprodukte
- DIN EN ISO 10993-1 Biologische Beurteilung von Medizinprodukten – Teil 1: Beurteilung und Prüfungen im Rahmen eines Risikomanagementsystems
Comments are closed.