Risikomanagement für Medizinprodukte – Missverständnisse um den Einsatz der FMEA22 | 09 | 21

Etwas mehr als zwei Jahre ist es her, dass mir bei einer Schulung zu normativen Anforderungen und dazu konformer Umsetzung eines Risikomanagementsystems für Medizinprodukte gemäß der internationalen und harmonisierten Norm EN ISO 14971:2012 erstmals eine recht kategorisch vorgetragene Behauptung begegnete, die sich so hartnäckig fest gesetzt hat, wie ich sie persönlich für falsch halte. Sie begegnet mir seitdem bis heute des Öfteren in unterschiedlichen Ausprägungen und sie erzeugt viel Verunsicherung und fragende Blicke.
Verbotene Methode?
Es geht um die Anwendung der FMEA-Methode zur Bewertung von Gefährdungen für Patienten sowie Anwender von Medizinprodukten über den gesamten Produktlebenszyklus von der Entwicklung bis zur sachgerechten Entsorgung und Vernichtung.
„Benutzen Sie nicht die FMEA zur Bewertung von Risiken!“ hieß seinerzeit die These des Referenten, die vielfach als Verbot dieser Methode missverstanden wird.
Perfekt wurde die Verwirrung mit Behandlung der Inhalte von Anhang D der EN ISO 14971:2012. Dieser (nicht normative sondern informative!) Anhang beschreibt grundsätzliche auf Medizinprodukte anzuwendende Risikokonzepte, an denen sich der Anwender bei der Ausgestaltung seines Systems zur Bewertung von potenziellen Gefährdungen und Gefährdungssituationen orientieren kann. Die methodischen Ähnlichkeiten zu einer FMEA, wie in DIN EN 60812 definiert, sind doch nicht von der Hand zu weisen, wozu dieser kategorische Ausschluss des Referenten?
Mit der Methode an sich war diese Meinung des Referenten nicht zu begründen, der Knackpunkt musste woanders gesucht werden.
Same, same but different: Knackpunkt Risikoprioritätszahl
Nach der Schulung ist vor der Arbeit und so wurde mir die grundlegende Renovierung des Risikomanagementprozesses unserer Organisation in die Hand gelegt. Mit dem Aufbau unserer produktspezifischen Risikomanagementakten wurden auch eine Risikopolitik und normkonforme Bewertungskriterien etabliert, die sowohl ein Abschätzen der Schwere des potenziellen Schadens (S) als auch dessen Wahrscheinlichkeit des Eintretens (W) ermöglichen.
Beide Dimensionen sind Bestandteil der aus der Methodik für Prozess-FMEA bekannten Risikoprioritätszahl (RPZ), die dazu mit der dritten Dimension, der Entdeckungswahrscheinlichkeit (E) anhand der Formel [S x W x E = RPZ] verknüpft werden. Den Dimensionen werden Zahlenwerte zwischen 1 und 10 zugeordnet, sodass sich je Risiko Werte im Bereich zwischen 1 und 1000 ergeben, die ab einem definierten Schwellenwert Maßnahmen zur Fehlervermeidung erforderlich machen.
Von diesem Bewertungskonzept sollte sich lösen, wer die FMEA-Methodik zur normkonformen Bewertung von Risiken für Medizinprodukte verwendet, da hierbei die Dimensionen nicht gewichtet werden. So könnte ein katastrophaler Schaden, der sich bei der Anwendung von Medizinprodukten meist durch schwerwiegende Gesundheitsschädigung oder Tod des Patienten auswirkt, kombiniert mit niedriger Auftretenswahrscheinlichkeit und hoher Entdeckbarkeit dazu führen, dass das Risiko dieses potenziell tödlichen Ereignisses als akzeptabel bewertet werden könnte.
Durch fehlende Gewichtung der Dimensionen sind die Faktoren zudem austauschbar und erzeugen in verschiedenen Kombinationen die gleiche RPZ. Dieses Problem wird auch nicht dadurch gelöst, dass die Entdeckungswahrscheinlichkeit nur in dem Maße in die Betrachtung einfließt, in dem die Entdeckung vorbeugende Maßnahmen im Rahmen der ISO 14971:2019 ermöglicht. Da dies für unsere Anwendungen nicht der Fall ist, wird auf die Einbeziehung der Entdeckungswahrscheinlichkeit grundsätzlich verzichtet.
Ein möglicher Ausweg
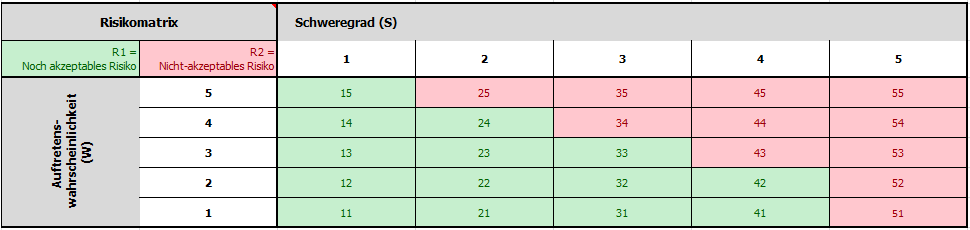
…ist die geschickte Definition der Wertebereiche für die halbquantitative Wahrscheinlichkeitsachse und der qualitativen Schweregradachse für die verwendete Risikomatrix im Risikomanagementplan, die wie im folgenden Beispiel gewählt werden können:

Die Niveau-Achsen werden nachfolgend in der Risikoakzeptanzmatrix gewichtet dargestellt. Hierbei kommen die RPZ jeweils nur einmal in der Matrix vor und ergeben somit ein klares Bewertungsergebnis hinsichtlich der Akzeptanz des potenziellen Risikos.
Fazit
Spätestens mit der Veröffentlichung des ISO/TR 24971:2020, der als Handlungsleitfaden zur Umsetzung der EN ISO 14971:2019 publiziert wurde, sollte sich das Missverständnis um ein FMEA-Verbot eigentlich erledigt haben, da in Anhang B „Techniques that support risk analysis“ unter anderem auch explizit die FMEA als hilfreich aufgeführt wird.
Referenzen:
EN ISO 14971:2019
ISO/TR 24971:2020
EN ISO 60812:2005
Thematische Verknüpfung: https://blog.dgq.de/ein-restrisiko-bleibt-immer-bestehen-risikomanagement-fuer-medizinprodukte/
DGQ-Weiterbildungsangebote rund um Qualitätsmanagement für Medizinprodukte sowie der FMEA-Methode
Im DGQ-Lehrgang „DIN EN ISO 13485 – Qualitätsmanagement für Medizinprodukte“ eignen Sie sich umfangreiches Wissen zu einem Managementsystem nach DIN EN ISO 13485 an. Sie sind auf dem aktuellen Stand der nationalen und europäischen Gesetzgebung. Sie lernen die zusätzlichen Anforderungen von DIN EN ISO 13485 gegenüber DIN EN ISO 9001 kennen. Mit diesem Lehrgang sind Sie bereit, ein Managementsystem nach ISO 13485 einzuführen, aufrechtzuerhalten und zu verbessern. Jetzt anmelden »
Verstehen Sie anhand des Trainings „Risikomanagement für Medizinprodukte“ den Sinn eines Risikomanagementsystems, die Vernetzung mit Kernprozessen des Qualitätsmanagementsystems und eignen sich anhand praktischer Übungen die Durchführung einer Risikoanalyse an. Sie sind in der Lage, Risiken zu bewerten und Maßnahmen abzuleiten, um Risiken zu mindern, sowie Risiko-Nutzen-Analysen durchzuführen. Zudem lernen Sie die aktuellen regulatorischen Anforderungen an das Risikomanagement für Medizinprodukte nach DIN EN ISO 14971 in Europa sowie vergleichsweise in den USA kennen. Jetzt anmelden »
Im FMEA-Basistraining erlernen Sie das Basiswissen der FMEA-Methode und machen sich mit ihrer praktischen Anwendung vertraut. Sie erlernen, wie Sie die FMEA zielgerichtet im Rahmen von Produktentwicklungen zur Risikoabsicherung sowie im Rahmen der Produktions- und Montageplanung zur Prozessoptimierung einsetzen können. Jetzt anmelden »
Comments are closed.